
Proteomic analysis using quadrupole ion mobility time-of-flight mass spectrometry
Direct analysis of high-resolution data: non-targeted proteomics analysis using quadrupole ion mobility time-of-flight mass spectrometry
Keith Richardson 1 , Christopher J. Hughes 1 , Arkadiusz Grzyb 1 , Dominic Helm 2 , Bernhard Kuster 2 , Jason Wildgoose 1
1 Waters Corporation (Manchester, UK)
2 Technical University of Munich (Germany Freising)
Application advantage
â– Improve protein recognition and increase proteome coverage
â– Reliable identification even at very low concentrations
â– Efficient LC/MS/MS analysis to speed decision making
Introduction
As the complexity of bottom-up proteomic analysis continues to increase, the ability of mass spectrometers is challenged, requiring more detailed information to meet user needs. In recent years, the accuracy, sensitivity, and mass accuracy of mass spectrometers have improved significantly, resulting in better data quality, improved peptide sequence labeling, and more accurate identification.
As hardware performance improves, we have developed a series of new LC/MS acquisition schemes and fragmentation mechanisms, including parent ion discovery (PID) methods, non-information dependent acquisition (DIA), ion mobility (IM) assist methods, and Electron Transfer Dissociation (ETD). Currently, the IM analysis 1, can enhance the specificity of DIA collected, e.g. HDMS E are mainly used in various types of cross-sectional analysis and structural analysis thereof. 2
This study demonstrates the advantages of a new high-definition data acquisition (HD-DDA) model for protein and peptide identification, which combines ion mobility into a quadrupole time-of-flight mass spectrometer. HD-DDA uses a high duty cycle mode to facilitate decision making and enable high sensitivity and selectivity.
experiment
The cytoplasmic contents of E. coli and HeLa cells were digested with trypsin. The lysate was injected into a nanoACQUITY UPLC system equipped with an ACQUITY UPLC BEH 1.7 μm, 15 cm × 75 μm column coupled to a SYNAPT G2-Si mass spectrometer. Process and search data using ProteinLynx Global SERVER and/or Mascot 3 .
Results and discussion
HD-DDA improvements include enhanced broadband full support 4, which can signal increased by 5 to 10 times, and enhance the decision making logic may be switched between the MS and MS / MS mode. Broadband enhancement uses ion mobility to separate product ions in a single charge state, combined with repeller synchronization to achieve a near 100% duty cycle, as shown in Figure 1.
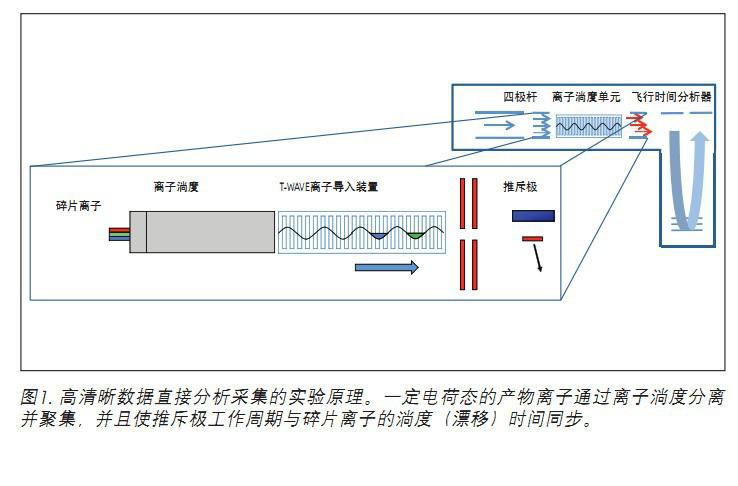
HD-DDA acquisition is typically performed in a non-targeted mode and can be supplemented by an unlimited target and/or exclusion list. The collision energy can be stepped, tilted, or determined in real time based on m/z and charge states. Data can be processed and searched through the ProteinLynx Global SERVER or using vendor-neutral search algorithms and verification tools, such as Mascot and Scaffold 5 .
The advantages of broadband enhancement are demonstrated in Figure 2. Among them, E. coli trypsin digest was analyzed by nanoliter LC/MS/MC. Data acquisition uses conventional DDA and HD-DDA, respectively. In both cases, 0.1 second MS/MS data for the same time in the LC peak was selected. In this experiment, after averaging the entire MS/MS spectrum, the signal was enhanced by a factor of five.
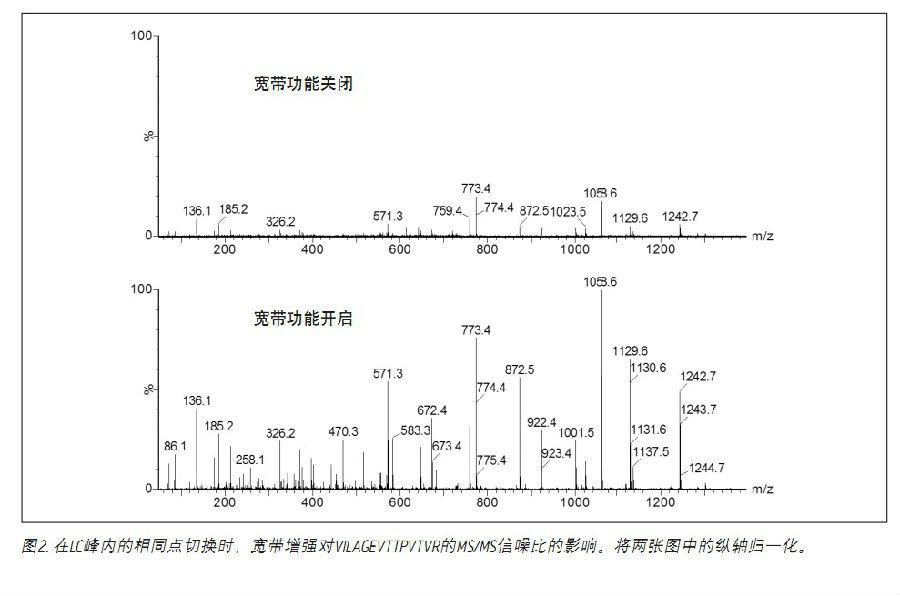
In this experiment, 15 parallel MS/MS experiments were performed per survey scan. As shown in Figure 3, the results show that the increase in MS/MS total ion current (TIC) is a function of the number of MS/MS channels when comparing DDA to HD-DDA. The average increase per run was 420%, which is consistent with the results shown in Figure 2. The inset is an example of 15 MS/MS channels, demonstrating that MS/MS data with good signal to noise ratios can be easily obtained from the lower abundance of peptides present in the sample.
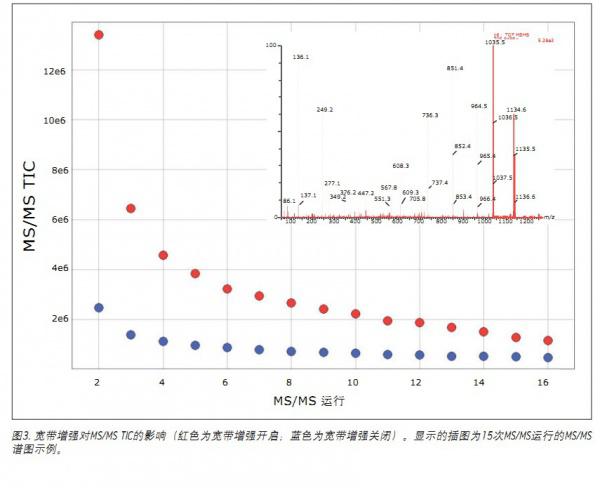
Figure 4 shows that bottom-up LC/MS proteomics experiments on the same E. coli samples using HD-DDA can increase sensitivity. Panel A shows an increase in the number of peptide sequence matches. The Wien intersection map in Figure B compares the number of protein identifications. A significant increase in the number of identified peptides (34.8%) and protein (42.8%) was observed.
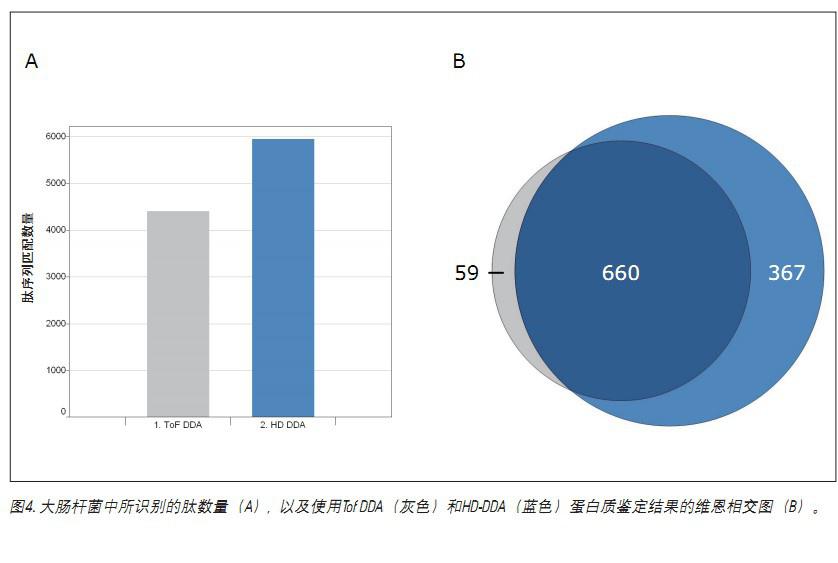
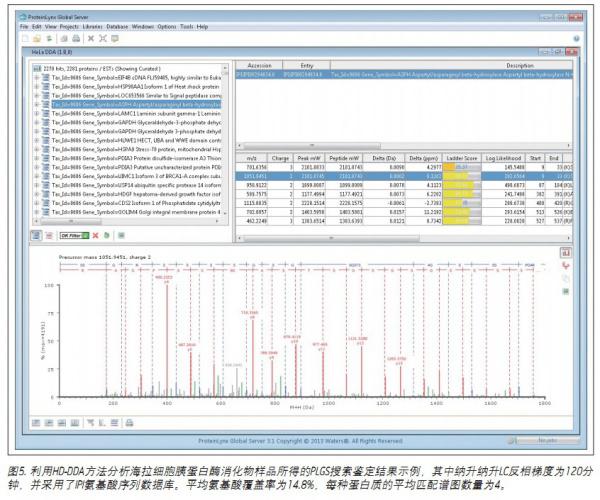
Figure 5 shows the search results for a more challenging sample. The PLGS search results for HeLa cell trypsin digest analysis are summarized here. A total of more than 2,200 proteins were identified as having an identification confidence threshold of over 95%. In this experiment, the spectrum identification rate was 38%.
in conclusion
â– HD-DDA broadband enhancement usually boosts signals by 5 to 10 times
â– The quality of the spectral data of low abundance substances/peptides has been significantly improved
â– The proportion of MS/MS spectra obtained correctly matched is greatly increased
â– Increased identification of HD-DDA data is a direct result of improved sensitivity and improved spectral quality
references
1. Giles K. Travelling wave ion mobility. Int J Ion Mobility Spectrom. 2013; 16(1): 1-3.
2. Rodriguez-Suarez E, Hughes C, Gethings L, Giles K, Wildgoose J, Stapels M, Fadgen K, Geromanos SJ, Vissers JPC, Elortza F, Langridge JI. An Ion Mobility Assisted Data Independent LC-MS Strategy for the Analysis Of Complex Biological Samples. Current Anal Chem. 2013 April; 9(2):199-211.
3. Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identi?cation by searching sequence databases using mass spectrometry data. Electrophoresis. 1999 Dec; 20(18):3551-67.
4. Wildgoose, et. al. A comparison of methods of improving the duty cycle on orthogonal TOF mass analyzers. ASMS 2005.
5. Searle BC. Scaffold: a bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics. 2010 Mar; 10(6): 1265-9.
To download a clear and complete PDF document please click: http://?lid=134743944&cid=511436
Canned Tuna In Vegetable Oil,Jack Mackerel Canned,Canned Tuna In Oil,Canned Tuna Chunk
DOCANNED , https://www.docannedfamily.com